
OVERALL SAFETY SUMMARY
In healthy subjects and hospitalized patients with PCR-confirmed SARS-CoV-2 infection, graded elevations in ALT and AST have been observed with a loading dose of remdesivir 200 mg administered intravenously on Day 1 followed by 100 mg administered intravenously once daily for up to 9 days. The mechanism of these elevations is unknown.
Patients should have appropriate clinical and laboratory monitoring to aid in the early detection of any potential adverse events. The decision to continue or discontinue remdesivir after the development of an adverse event should be made based on the clinical risk-benefit assessment for the individual.
Clinical Trials Experience
Clinical Trials Experience in Subjects with COVID-19
NIAID ACTT-1 was a randomized, double-blind, placebo-controlled clinical trial in hospitalized adult subjects with mild, moderate, and severe COVID-19 treated with remdesivir (n=532) or placebo (n=516) for up to 10 days. Subjects treated with remdesivir received 200 mg on Day 1 and 100 mg once daily on subsequent days. The collection of adverse event data in this trial was limited to severe (Grade 3) or potentially life-threatening (Grade 4) adverse events, serious adverse events, adverse events leading to study drug discontinuation, and moderate (Grade 2) severity or higher hypersensitivity reactions. Rates of adverse reactions (≥ Grade 3), serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 6.
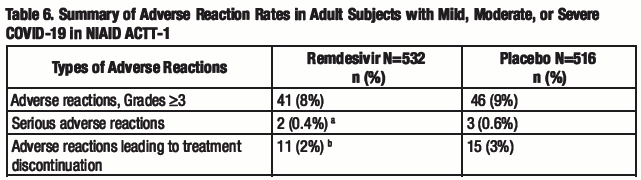
a.Seizure (n=1), infusion-related reaction (n=1).
b.Seizure (n=1), infusion-related reaction (n=1), transaminases increased
Study GS-US-540-5773 was a randomized, open-label clinical trial in hospitalized adult subjects with severe COVID-19 treated with remdesivir 200 mg on Day 1 and 100 mg once daily for 5 (n=200) or 10 days (n= 197). Adverse reactions were reported in 33 (17%) subjects in the 5-day group and 40 (20%) subjects in the 10-day group. The most common adverse reactions occurring in at least 5% of subjects in either tile remdesivir 5-day or 10-day group, respectively, were nausea (5% vs 3%), AST increased (3% vs 6%), and ALT increased (2% vs 7%). Rates of any adverse reaction, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 7.
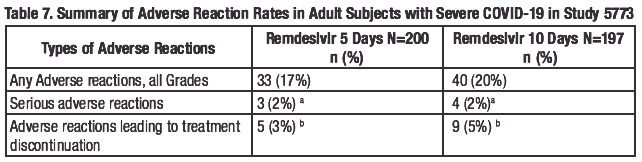
a.Transaminases increased (n=5), hepatic enzyme increased (n=1), hypertransaminasaemia (n=1).
b. Transaminases increased (n=4), hepatic enzyme increased (n=2), LFT increased (n=2),
hypertransaminasaemia (n= 1 ), ALT increased (n= 1), ALT increased and AST increased (n=2), injection
site erythema (n=1), rash (n=1).
Study GS-US-540-5774 was a randomized, open-label clinical trial in hospitalized adult subjects with moderate COVID-19 treated with remdesivir 200 mg on Day 1 and 100 mg daily for 5 (n=191) or 10 days (n=193), or standard of care (SOC) only (n=200). Adverse reactions were reported in 36 (19%) subjects in the 5-day group and 25 (13%) subjects in tile 10-day group. The most common adverse reaction occurring in at least 5% of subjects in the remdesivir groups was nausea (7% in the 5-day group, 4% in the 10-day group). Rates of any adverse reaction, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 8.
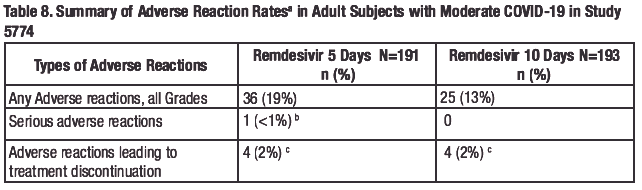
a. Attribution of events to study drug was not performed for the SOC group.
b. Heart rate decreased
c. ALT increased (n=2), ALT increased and AST increased (n=1 ), hypertransaminasaemia (n=1 ), blood
alkaline phosphatase increased (n=1), rash (n=2), heart rate decreased (n=1)
Less Common Adverse Reactions
Clinically significant adverse reactions that were reported in <2% of adult subjects exposed to remdesivir in clinical trials are listed below :
- Hypersensitivity reactions [see Warnings and Precautions].
- Generalized seizure
- Rash
Laboratory Abnormalities
Study GS-US-399-5505 was a Phase 1, randomized, blinded, placebo-controlled clinical trial in healthy adult volunteers administered remdesivir 200 mg on Day 1 and 100 mg for either 4 days or 9 days. Mild (Grade 1, n=8) to moderate (Grade 2, n=1) elevations in ALT were observed in 9 of 20 subjects receiving 1 O days of remdesivir; the elevations in ALT resolved upon discontinuation of remdesivir. No subjects (0 of 9) who received 5 days of remdesivir had graded increases in ALT.
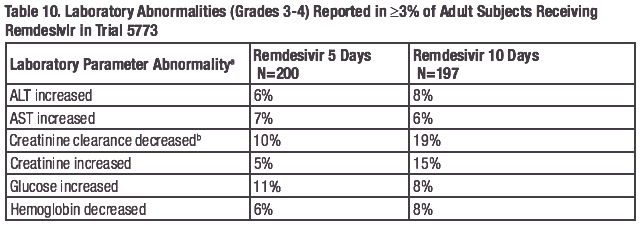
The frequencies of laboratory abnormalities (Grades 3-4) occurring in at least 3% of adult subjects with COVID-19 receiving remdesivir in Trials NIAID ACTT-1, 5773, and 5774 are presented in Table 9, Table 10, and Table 11, respectively.
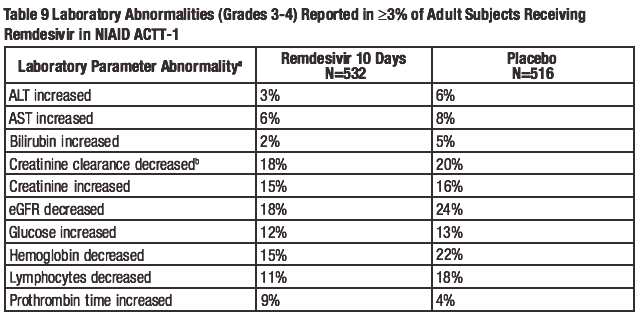
a.
Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS
(DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July
2017.
b. Based on the Cockcroft-Gault formula.
a.
Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS
(DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July
2017.
b. Based on the Cockcroft-Gault formula.
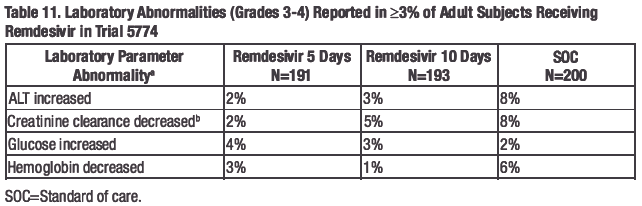
a.
Frequencies are based on treatment-emergent laboratory abnormalities. Graded per Division of AIDS
(DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.1 dated July
2017.
b. Based on the Cockcroft-Gault formula.